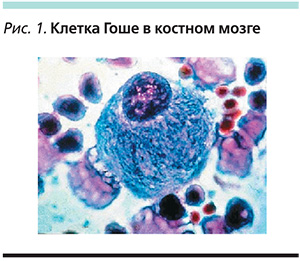
Болезнь Гоше (БГ) относится к группе лизосомных болезней накопления. Частота БГ в общей популяции –1 на 40–75 тыс. населения. Среди евреев-ашкенази (выходцев из Восточной Европы) частота встречаемости этого заболевания наиболее высока: до 1 на 450 человек. Впервые это заболевание было описано французским дерматологом Ф.Ч. Гоше в 1882 г. В основе БГ лежит дефект фермента β-D-глюкозидазы, который находится внутри лизосом и отвечает за расщепление сложного липида – глюкоцереброзида на глюкозу и церамид. Вследствие недостаточной активности β-D-глюкозидазы глюкоцереброзид не расщепляется полностью и накапливается в макрофагах. «Нагруженные» липидами клетки, называемые клетками Гоше, являются патологическим субстратом болезни (рис. 1). Инфильтрация клетками Гоше различных органов и тканей, в которых присутствуют макрофаги, объясняет полисистемный характер этой болезни: клетки Гоше накапливаются в печени, селезенке, костном мозге, легких, почках, лимфатических узлах [1].
БГ наследуется по аутосомно-рецессивному типу. В настоящее время описано более 300 мутантных аллелей, частично или полностью блокирующих каталитическую активность фермента β-D-глюкозидазы [2]. Ген β-глюкоцереброзидазы картирован на хромосоме (1q21).
Классификация
В зависимости от клинического течения выделяют три типа БГ, основанных на наличии или отсутствии симптомов поражения центральной нервной системы (ЦНС). В частности, 1-й тип БГ характеризуется отсутствием неврологической патологии, он встречается чаще других (более 90 % всех случаев болезни). Два других типа БГ ассоциируются с вовлечением в процесс ЦНС: 2-й тип – острый невропатический, или инфантильный; 3-й – подострый невропатический [3].
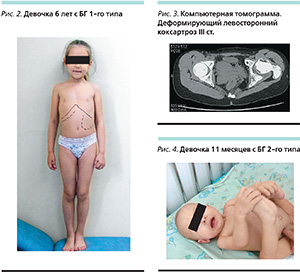
Клиническая картина 1-го типа БГ характеризуется нарастающим увеличением паренхиматозных органов (печени и селезенки), панцитопенией и развитием костной патологии [4]. Заболевание одинаково часто диагностируется среди лиц обоих полов и может манифестировать в любом возрасте. Течение хроническое, прогрессирующее. Отмечается отставание детей в физическом и половом развитии. Спленомегалия является постоянным и наиболее ранним признаком БГ, при пальпации селезенка имеет плотную консистенцию. Гепатомегалия при БГ выражена в меньшей, чем спленомегалия, степени и, как правило, развивается в более поздние сроки (рис. 2). Редкие симптомы включают инфильтраты в легких, легочную гипертензию, хроническую почечную и печеночную недостаточность, портальную гипертензию [5, 6]. Основная причина стойкой инвалидизации при БГ 1-го типа – поражение скелета. Изменения в костной ткани – следствие замещения нормальных элементов костного мозга инфильтратами клеток Гоше, сопровождаемое нарушением нормальных физиологических процессов. Обычно в первую очередь поражаются бедренные кости, затем другие трубчатые кости и позвоночник. У большинства больных БГ снижается минеральная плотность костной ткани и происходит потеря костной массы [7]. Накопление клеток Гоше в костном мозге приводит к отеку, увеличению внутрикостного давления и острым болевым ощущениям, известным как костные кризы. Клиническая картина костного криза подобна картине остеомиелита, но в отличие от последнего при костных кризах посевы крови на бактериальную флору отрицательны [8]. Прогрессирующая инфильтрация клетками Гоше костного мозга может приводить к сужению сосудов, их окклюзии, тромбозу, что вызывает развитие патологических очагов остеонекроза и склерозирование костной ткани [9]. Остеонекроз обычно поражает головки бедренных костей, но у некоторых больных патологическим изменениям в головке бедра может предшествовать вовлечение в процесс шейки бедра. Без своевременного лечения анатомические, функциональные и трофические нарушения в пораженной головке бедренной кости приводят к тяжелым вторичным изменениям структур сустава с развитием коксартроза (рис. 3). Ряду таких пациентов проводится оперативное лечение — эндопротезирование тазобедренного сустава, позволяющее восстанавливать опорно-двигательную функцию нижней конечности [10].
 При БГ 2-го типа возраст начала болезни – 1-й год жизни, хотя описана манифестация в виде водянки плода. Болезнь манифестирует гепатоспленомегалией, окуломоторными аномалиями и с 6 месяцев – прогрессирующей задержкой психомоторного развития (рис 4). Первыми симптомами, привлекающими внимание, часто являются нарушение глотания, поперхивание, осложняющиеся аспирационной пневмонией. У детей отмечаются также тризм, билатеральное фиксированное косоглазие, прогрессирующая спастичность с характерной для 2-го типа ретракцией шеи, гиперрефлексия, положительный симптом Бабинского и другие патологические рефлексы, потеря ранее приобретенных навыков. Тонико-клонические судорожные приступы, как правило, возникают на поздних стадиях болезни и резистентны к назначаемой противосудорожной терапии. Течение заболевания – быстропрогрессирующее с летальным исходом на 1–2-м годах жизни [11].
При БГ 2-го типа возраст начала болезни – 1-й год жизни, хотя описана манифестация в виде водянки плода. Болезнь манифестирует гепатоспленомегалией, окуломоторными аномалиями и с 6 месяцев – прогрессирующей задержкой психомоторного развития (рис 4). Первыми симптомами, привлекающими внимание, часто являются нарушение глотания, поперхивание, осложняющиеся аспирационной пневмонией. У детей отмечаются также тризм, билатеральное фиксированное косоглазие, прогрессирующая спастичность с характерной для 2-го типа ретракцией шеи, гиперрефлексия, положительный симптом Бабинского и другие патологические рефлексы, потеря ранее приобретенных навыков. Тонико-клонические судорожные приступы, как правило, возникают на поздних стадиях болезни и резистентны к назначаемой противосудорожной терапии. Течение заболевания – быстропрогрессирующее с летальным исходом на 1–2-м годах жизни [11].
Манифестация болезни при 3-м типе БГ – раннее детство или второе десятилетие (1-й месяц – 14 лет). Заболевание характеризуется более медленным прогрессированием. В дебюте – гепатоспленомегалия и неврологические симптомы, сходные с таковыми при 2-м типе БГ, но менее тяжело выраженные.
Самая ранняя неврологическая симптоматика проявляется окуломоторными расстройствами, включая супраорбитальную офтальмоплегию. Главным неврологическим маркером являются миоклонические судороги кортикальной природы: от быстрых подергиваний в нескольких группах мышц как в покое, так и при нагрузке, которые постепенно нарастают и делаются навязчивыми, прогрессируя в генерализованные тонические судороги. Интеллектуальные нарушения варьируются от незначительных изменений до тяжелой деменции. Возможно развитие мозжечковых нарушений, а также расстройств речи и письма, могут наблюдаться поведенческие изменения, эпизоды психоза. Срок летального исхода варьируется на фоне прогрессирующих неврологических расстройств. Продолжительность жизни больных при БГ 3-го типа составляет 12–17 лет, но описаны случаи 30–40-летней выживаемости [11].
Международные критерии диагностики
В соответствии с международными критериями в настоящее время диагностика БГ складывается из ряда последовательных этапов:
- обнаружение характерных клинических признаков заболевания;
- измерение активности β-D-глюкозидазы в лейкоцитах;
- молекулярно-генетический анализ [12].
Помимо специфической диагностики существуют определенные изменения, выявляемые рутинными лабораторно-инструментальными методами.
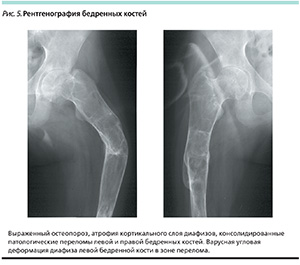
У большинства больных БГ отмечаются тромбоцитопения, лейкопения и анемия как проявления гиперспленизма. При ультразвуковом исследовании и магнитно-резонансной томографии печени и селезенки определяются очаги как с повышенной, так и с пониженной интенсивностью сигнала. Эти очаги являются зонами ишемии и фиброза из-за повышенной инфильтрации клетками Гоше. У пациентов с невропатическими типами БГ изменения, выявляемые при электроэнцефалографии (ЭЭГ), неспецифичны и чаще проявляются дезорганизованным паттерном ЭЭГ сна, дисфункцией корково-подкорковых взаимодействий, дисфункцией и раздражением срединных и подкорковых структур, формированием вспышек полиморфной эпилептиформной активности и пароксизмами острых полифазных потенциалов [13]. На рентгенограммах при БГ выявляют истончение надкостницы, эндостальную зубчатость и пониженную трабекулярность костной ткани. Дистальные метафизы бедра вздуваются в виде булавы или колбы, в литературе также известные как «колбочки Эрленмейера». Тяжесть поражения трубчатых костей различна – от классических деформаций метафизов трубчатых костей до тяжелых патологических переломов, очагов литической деструкции и асептических некрозов головок бедренных костей (рис. 5). Денситометрия показывает снижение минеральной плотности костной ткани и используется как для ранней диагностики системной остеопении, так и для мониторинга эффективности лечения.
Дифференциальный диагноз
При 1-м типе БГ в зависимости от вида манифестации дифференциальный диагноз должен проводиться с разнообразными экзогенными и наследственными заболеваниями, сопровождающимися висцеромегалией, острыми болями в костях, кровоточивостью (вирусный гепатит, остеомиелит, костный туберкулез, гемофилии, сфинголипидозы); при 2-м и 3-м типах БГ – со всеми инфантильными формами сфинголипидозов с гепатоспленомегалией (болезнь Ниман-Пика, типы А, С), GM1-ганглиозидозом, галактосиалидозом, болезнью Вольмана, болезнью Фарбера (атипичные формы), а также врожденной окуломоторной апраксией [14].
Лечение
БГ стала первым заболеванием, для которого была разработана патогенетическая ферментозаместительная терапия, являющаяся единственным эффективным методом лечения БГ, купирующим основные клинические проявления заболевания, улучшая качество жизни больных БГ и не оказывая выраженных побочных эффектов. Имиглюцераза (Церезим®) является аналогом β-D-глюкозидазы и производится с помощью ДНК-рекомбинантной технологии. Под действием имиглюцеразы происходит гидролиз гликолипида глюкоцереброзида до глюкозы и церамида по обычному пути метаболизма мембранных липидов. Церезим® показан для длительной заместительной ферментотерапии больных с подтвержденным диагнозом БГ без поражения нервной системы (1-й тип) или с хроническим поражением нервной системы (3-й тип), у которых имеются клинически значимые неневрологические проявления заболевания [15]. В связи с гетерогенностью БГ доза препарата для каждого пациента должна подбираться индивидуально и может повышаться или понижаться в зависимости от успешности достижения терапевтических целей на основании оценки клинических проявлений. Первоначальная доза имиглюцеразы составляет 30–60 ЕД/кг при 1-м типе БГ и 120 ЕД/кг при 3-м. Препарат вводится внутривенно капельно медленно 1 раз в 14 дней. Методы эффективной терапии для 2-го типа не описаны. Опубликованы данные по результатам ферментозаместительной терапии у больных, в течение 10 лет получавших алглюцеразу (Цередаза®) или имиглюцеразу (Церезим®). Продемонстрировано значительное устойчивое улучшение состояния пациентов с болезнью Гоше, которое оценивалось по таким параметрам, как уровень гемоглобина, тромбоцитов, объем печени и селезенки (у неспленэктомизированных больных), наличие костных болей и костных кризов [16].
 Комплексная терапия проявлений остеопороза направлена на замедление и прекращение потери костной массы, повышение ее прочности, предотвращение переломов костей и включает применение бисфосфонатов, альфакальцидола, солей кальция, оссеин-гидроксиапатитного соединения. Симптоматическая терапия скелетных осложнений при БГ: анальгетики во время костных кризов, антибактериальная терапия. При хирургических вмешательствах существует повышенный риск кровотечения и инфицирования.
Комплексная терапия проявлений остеопороза направлена на замедление и прекращение потери костной массы, повышение ее прочности, предотвращение переломов костей и включает применение бисфосфонатов, альфакальцидола, солей кальция, оссеин-гидроксиапатитного соединения. Симптоматическая терапия скелетных осложнений при БГ: анальгетики во время костных кризов, антибактериальная терапия. При хирургических вмешательствах существует повышенный риск кровотечения и инфицирования.
Контроль течения заболевания у детей на фоне терапии проводят в соответствии с рекомендациями по минимально необходимому мониторингу состояния больных при БГ, разработанными Объединенной международной группой по изучению БГ (ICGG). При этом контроль анализов крови необходимо проводить один раз в 3 месяца, контроль размеров паренхиматозных органов (по данным ультразвукового исследования, магнитно-резонансной томографии) один раз в 6 месяцев, а также при изменении дозировки или значительных клинических осложнениях.
Контроль состояния костной ткани проводят один раз в год.
Особую роль при проведении мониторинга в процессе патогенетического лечения БГ приобретает определение активности хитотриозидазы, которая синтезируется макрофагами и резко повышена (в 100 и более раз) у большинства больных БГ. На фоне адекватно подобранной ферментозаместительной терапии активность хитотриозидазы значительно снижается, что является одним из важных критериев оценки ее эффективности. Контроль активности хитотриозидазы должен проводиться 1 раз в 4 месяца [12].
Ошибки и необоснованные назначения
Проведение спленэктомии с лечебной целью противопоказано (за исключением единичных, редких случаев специальных показаний). После удаления селезенки клетки Гоше в основном накапливаются в печени, костях, легких. Нарушение структуры и функции этих органов вследствие «использования не по назначению» приводит к необратимым последствиям: развитию цирроза печени, деформации костей и суставов, фиброзу легких и тяжелой легочно-сердечной недостаточности. Среди больных, перенесших спленэктомию, отмечена также ассоциация между повышенным содержанием тромбоцитов в крови и остеонекрозом, что может быть связано с развитием внутрикостного тромбоза [9].
При доказанном диагнозе БГ не нужны повторные пункции костного мозга и другие инвазивные диагностические мероприятия (биопсия печени, селезенки).
Крайне опасно оперативное лечение костных кризов, которые ошибочно рассматриваются как проявления остеомиелита. Лечение должно быть консервативным (покой, обезболивающие препараты, наблюдение). Решение о необходимости оперативных мероприятий должны принимать специалисты, имеющие опыт ведения и лечения пациентов с БГ.
 При БГ противопоказано назначение кортикостероидов (преднизолона и других препаратов) с целью купирования цитопенического синдрома.
При БГ противопоказано назначение кортикостероидов (преднизолона и других препаратов) с целью купирования цитопенического синдрома.
Необоснованно назначение препаратов железа пациентам с развернутой картиной БГ, т. к. малокровие в этих случаях носит характер «анемии воспаления» и требует лечения основного заболевания, т. е. назначения заместительной ферментной терапии.
Профилактика
Пренатальную диагностику можно осуществлять с помощью определения активности β-D-глюкозидазы в культуре амниоцитов, биоптате и культуре хориона, а также методами ДНК-диагностики.
